Koronnym argumentem środowisk antyszczepionkowych jest twierdzenie, że program szczepień przeciwko COVID-19 to eksperyment medyczny. Informacja ta jest powtarzana jak mantra nie tylko przez zwykłych internautów, ale przede wszystkim przez liderów tej społeczności. Co zdumiewające, nawet tych mogących pochwalić się wykształceniem medycznym. Z takim twierdzeniem często idzie w parze przeświadczenie, że szczepionki nie przeszły wszystkich wymaganych faz badań klinicznych, a ich dopuszczenie odbyło się w trybie awaryjnym. Podnoszony jest również argument, jakoby ani producent, ani władze nie ponosiły odpowiedzialności za działania niepożądane szczepionek.

Rozpowszechnianie takich informacji świadczy o nieznajomości regulacji prawnych i podstawowych zasad przeprowadzania badań klinicznych. Stosowanie szczepionki dopuszczonej do obrotu w sposób określony w pozwoleniu nie jest eksperymentem medycznym. Co więcej, szczepionki przeciwko COVID-19 nie zostały dopuszczone do obrotu w trybie awaryjnym, a w warunkowym, co gwarantuje że ich bezpieczeństwo jest ściśle monitorowane. Pacjenci natomiast nie są wcale pozbawieni możliwości ubiegania się o rekompensatę w przypadku wystąpienia działań niepożądanych.
Czym właściwie jest eksperyment medyczny?
W myśl ustawy o zawodach lekarza i dentysty badanie kliniczne zalicza się do eksperymentów medycznych. Ustawa wyróżnia dwa ich rodzaje:
- eksperyment leczniczy – jest wprowadzeniem nowych albo tylko częściowo wypróbowanych metod diagnostycznych, leczniczych lub profilaktycznych w celu osiągnięcia bezpośredniej korzyści dla zdrowia osoby chorej. Może on być przeprowadzony, jeżeli dotychczas stosowane metody nie są skuteczne albo jeżeli ich skuteczność nie jest wystarczająca. Nieraz stanowi on jedyną szansę na wyleczenie pacjenta.
- eksperyment badawczy – ma na celu przede wszystkim rozszerzenie wiedzy medycznej. Może być on przeprowadzany zarówno na osobie chorej, jak i zdrowej. Przeprowadzenie eksperymentu badawczego jest dopuszczalne, gdy uczestnictwo w nim nie jest związane z ryzykiem albo też ryzyko jest minimalne i nie pozostaje w dysproporcji do możliwych pozytywnych rezultatów takiego eksperymentu.
Z punktu widzenia powyższych definicji, badania kliniczne są rodzajem eksperymentu badawczego.
Zgoda uczestnika na udział w eksperymencie medycznym
Eksperyment medyczny może być przeprowadzony dopiero po uzyskaniu świadomej zgody jego uczestnika. Wyjątkiem jest tu szczególna sytuacja niecierpiąca zwłoki jak np. zagrożenie życia pacjenta. W takim przypadku dopuszcza się przeprowadzenie jedynie eksperymentu leczniczego, jednak aby móc to zrobić musi być jednocześnie spełniony szereg innych warunków. Zgodę na udział w eksperymencie medycznym uczestnik może wycofać w dowolnie wybranym momencie, bez podania przyczyny i bez negatywnych konsekwencji prawnych. Jeśli uczestnikiem badania jest osoba małoletnia poniżej 13 roku życia, zgody udziela jej przedstawiciel ustawowy. Jeśli ukończyła 13 rok życia, wtedy oprócz przedstawiciela ustawowego, zgodę musi wyrazić osoba małoletnia. Jeżeli np. rodzice nie mogą osiągnąć porozumienia w kwestii wyrażenia zgody, sprawę rozstrzyga sąd opiekuńczy.

Eksperyment medyczny a ubezpieczenie
Podmiot prowadzący eksperyment medyczny jest zobowiązany do zawarcia umowy ubezpieczenia OC na rzecz jego uczestników. Wyjątkiem jest tutaj sytuacja niecierpiąca zwłoki, kiedy występuje zagrożenie życia uczestnika eksperymentu leczniczego (nie stosuje się tego wyjątku w odniesieniu do eksperymentu badawczego).
Komisja bioetyczna
Eksperyment medyczny może być przeprowadzony wyłącznie po wyrażeniu pozytywnej opinii o projekcie przez niezależną komisję bioetyczną. Wniosek kierowany do komisji musi w szczegółowy sposób opisywać planowany eksperyment (m.in. liczbę jego uczestników, miejsce i czas wykonywania, planowane procedury, warunki włączenia i wykluczenia uczestnika z eksperymentu). Należy do niego załączyć również informację o warunkach ubezpieczenia uczestników. Komisja ocenia m.in. zasadność, wykonalność i plan badania, analizę przewidywanych korzyści i ryzyka, poprawność wyboru członków zespołu badawczego, jakość ośrodka, poprawność protokołu badania, zasady rekrutacji uczestników oraz wysokość odszkodowania i zawartą umowę OC.
Badanie kliniczne
Badanie kliniczne jest rodzajem eksperymentu badawczego, ale o ściśle określonych regułach i zasadach przeprowadzania. Są one określane mianem Dobrej Praktyki Klinicznej i opisują wymagania dotyczące zarówno przeprowadzania samych badań jak i ich monitorowania i raportowania.
Rola Urzędu Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Do przeprowadzenia badania klinicznego, oprócz pozytywnej opinii komisji bioetycznej, wymagane jest także pozwolenie Prezesa Urzędu Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych. Wniosek o uzyskanie zgody Prezesa Urzędu musi zawierać szereg dokumentów ściśle wymienionych w art. 37m. 2. prawa farmaceutycznego. Dokumentacja może być przedstawiona w języku angielskim, poza dokumentami przeznaczonymi do wiadomości uczestników badania (informacje dla pacjenta i formularz świadomej zgody). Jeżeli wniosek nie zawiera wszystkich wymaganych dokumentów i nie zostaną one uzupełnione w odpowiednim terminie, pozostaje on bez rozpoznania. Po otrzymaniu informacji o rozpoczęciu badania klinicznego, Prezes Urzędu wpisuje je do Centralnej Ewidencji Badań Klinicznych, a jego prawidłowy przebieg jest kontrolowany przez Inspekcję Badań Klinicznych.
Decyzja Prezesa Urzędu i opinia Komisji Bioetycznej są wydawane niezależnie od siebie. Rozpoczęcie badania klinicznego nie może odbyć się jednak przy braku którejkolwiek z nich:
Warunkiem rozpoczęcia badania klinicznego jest uzyskanie przez sponsora pozwolenia na rozpoczęcie badania klinicznego z dwóch niezależnych źródeł: komisji etycznej i Prezesa Urzędu. Dlatego badanie kliniczne można rozpocząć, jeżeli komisja bioetyczna wydała pozytywną opinię w sprawie prowadzenia badania oraz Prezes Urzędu wydał pozwolenie na prowadzenie badania klinicznego. (Grzegorz Cessak, Prezes Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych)
Dobra Praktyka Kliniczna (GCP, Good Clinical Practice)
Zasady Dobrej Praktyki Klinicznej (GCP) stanowią międzynarodowy standard etyczny i naukowy w zakresie prowadzenia badań klinicznych prowadzonych z udziałem ludzi. Gwarantują one ochronę praw, bezpieczeństwo uczestników tych badań oraz wiarygodność ich wyników. Określają one jednolity standard prowadzenia badań w krajach UE, Japonii, USA, Kanadzie i Szwajcarii. Obecny ich kształt jest efektem ustaleń Międzynarodowej Komisji ds. Harmonizacji (ICH, InternationalConference on Harmonisation) z 1995 roku oraz ich aktualizacji z 1996 i 2006 roku. W 1998 roku zostały po raz pierwszy wydane w języku polskim.
Fundamentalną zasadą GCP jest nadrzędna wartość prawa, bezpieczeństwa, zdrowia i dobra uczestników badania klinicznego, w stosunku do interesu nauki oraz społeczeństwa. Dodatkowo korzyści terapeutyczne jakie odniesie uczestnik badania, muszą przewyższać możliwe ryzyko związane z użyciem produktu leczniczego. Bezpieczeństwo pacjentów według zasad GCP postrzegane jest jako bezpieczeństwo indywidualne uczestników badania oraz potencjalnych, przyszłych jego beneficjentów.
Dzięki przyjęciu przez wspomniane kraje jednolitych zasad prowadzenia badań klinicznych, umożliwione zostało wzajemne uznanie uzyskanych przez te kraje danych. To z kolei pozwoliło na wprowadzenie nowych terapii na wiele rynków bez konieczności powielania eksperymentów medycznych.
Fazy badań klinicznych
Badania kliniczne są poprzedzone badaniami podstawowymi, które mają na celu m.in. określenie podstawy działania nowego produktu leczniczego, jego metabolizmu, farmakokinetyki i danych toksykologicznych. Przed rozpoczęciem badań z udziałem pacjentów potencjalny lek testuje się wcześniej w warunkach laboratoryjnych in vitro i in vivo z wykorzystaniem m.in. hodowli komórkowych i modeli zwierzęcych. Są to tzw. badania przedkliniczne. Na tym etapie zbierane są dane farmakologiczne, farmakokinetyczne, farmakodynamiczne, metaboliczne, toksykologiczne oraz, w przypadku szczepionki, dane odnośnie jej immunogenności. Badany jest również m.in. wpływ produktu na układ rozrodczy i potencjalne działanie kancerogenne. Dopiero po osiągnięciu obiecujących wyników badań przedklinicznych preparat może być zastosowany w badaniach klinicznych.
W badaniach klinicznych szczepionek porównuje się bezpieczeństwo i skuteczność nowej szczepionki w stosunku do nieaktywnego placebo lub kontroli, którą może być inna szczepionka przeciwko tej samej chorobie. Jeśli istnieje taka szczepionka, najczęściej z powodów etycznych, stosuje się badania porównawcze z istniejącym już produktem.
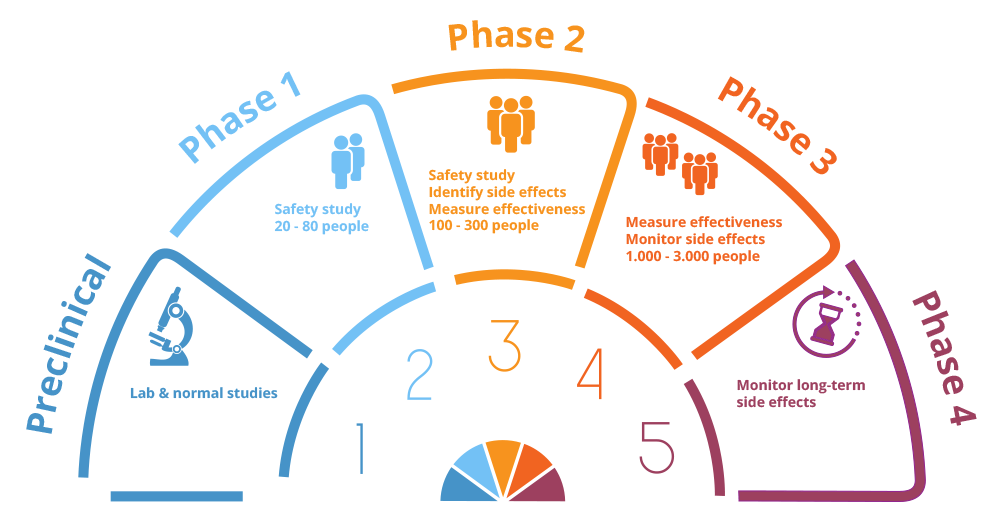
Teoretycznie badania te podzielone są na 4 fazy, jednak często różne fazy przebiegają niemal równolegle. Zdarza się również, że nie są prowadzone w kolejności od fazy I do IV.
Faza I – na tym etapie potencjalny lek zostaje po raz pierwszy zastosowany u ludzi. Ma on na celu ustalenie farmakokinetyki i bezpieczeństwa przyszłego leku. W przypadku szczepionki dokonuje się oceny efektów immunologicznych i tolerancji różnych dawek przez organizm. W tej fazie uczestniczy stosunkowo niewielka liczba osób, zwykle kilkadziesiąt.
Faza II – ma na celu uzyskanie dalszych danych dotyczących bezpieczeństwa, ale też po raz pierwszy oceniana jest skuteczność nowego produktu leczniczego. W tej fazie zazwyczaj określana jest optymalna dawka i potrzeba stosowania dawek przypominających i najkorzystniejszych odstępów czasowych między nimi. W fazie drugiej bierze udział zwykle kilkaset uczestników.
Faza III – na tym etapie potwierdzana jest skuteczność i bezpieczeństwo preparatu w większej populacji pacjentów (wielkość określona wymogami statystycznymi, z reguły kilka lub nawet kilkadziesiąt tysięcy). Badana populacja jest bardziej zróżnicowana i zbliżona do tej spotykanej w codziennej praktyce lekarskiej.
Badania kliniczne szczepionek różnią się nieznacznie od badań tradycyjnych leków. Główną przyczyną jest to, że szczepionki są przede wszystkim przeznaczone dla zdrowych osób jako środek zapobiegawczy, natomiast leki przeznaczone przede wszystkim do leczenia pacjentów z istniejącymi wcześniej chorobami. Stąd we wszystkich fazach badań klinicznych szczepionek biorą udział osoby zdrowe (chyba że prowadzone jest specjalne badanie dotyczące np. wytwarzania odporności poszczepiennej u osób z HIV). W badaniach klinicznych leków, najczęściej zdrowi pacjenci uczestniczą tylko w I fazie. Dopiero w II i III fazie potencjalny lek jest stosowany u chorych na konkretną chorobę, w której oczekujemy efektu terapeutycznego badanej substancji. W badania szczepionek angażuje się też większą liczbę uczestników. Im większa jest to liczba, tym ocena bezpieczeństwa szczepionki jest dokładniejsza, gdyż zwiększa się prawdopodobieństwo wykrycia rzadkich działań niepożądanych.
Zwieńczeniem badań fazy III jest przygotowanie wniosku o dopuszczenie produktu leczniczego do obrotu. Szczegółowe wyniki badań I-III fazy stanowią integralną część dokumentacji rejestracyjnej. Tekst Charakterystyki Produktu Leczniczego (ChPL), oraz tekst ulotki informacyjnej dla pacjentów dołączanej do opakowania, opracowane są na podstawie wyników tych trzech faz.
Faza IV – porejestracyjna
Po dopuszczenie na rynek rozpoczyna się wieloletni okres stosowania produktu leczniczego w codziennej praktyce medycznej. Dopiero wtedy można uzyskać informacje dotyczące jego skuteczności i bezpieczeństwa w ogólnej populacji. Dlatego badania te są kontynuowane po uzyskaniu dopuszczenia do obrotu. Jest to tzw. IV faza badań klinicznych, zwana inaczej porejestracyjną, w której badany lek jest stosowany zgodnie z warunkami określonymi w pozwoleniu na dopuszczenie do obrotu. Produkt leczniczy od pierwszego zastosowania u człowieka, aż do dnia zakończenia jego produkcji i stosowania, jest stale monitorowany. W tym sensie faza IV trwa tak długo, jak produkt jest dostępny na rynku. Prowadzona jest ciągła ocena skuteczności produktu oraz bezpieczeństwa w ramach porejestracyjnego systemu monitorowania działań niepożądanych. W fazie IV można też zgromadzić informacje służące do wykazania potencjalnego szerszego zastosowania niż określono początkowo. Jeśli natomiast zachodzi potrzeba przeprowadzenia badania dla nowych wskazań, postaci, czy dawek leku już obecnego na rynku, przeprowadza się kolejne badania fazy I-III.
Czy w związku z tym Narodowy Program Szczepień to eksperyment medyczny?
Jak podaje Strus-Wołos i Wołos Kancelaria Adwokacka s.c.:
Od chwili zatwierdzenia przez Komisję Europejską dopuszczenia szczepionki do obrotu, choćby warunkowego, jej stosowanie w żadnym wypadku nie może być uważane za eksperyment medyczny w rozumieniu art. 21 u.z.l.
W innym poście Strus-Wołos i Wołos Kancelaria Adwokacka s.c. zaznacza:
Trzeba odróżnić podawanie szczepionki po jej dopuszczeniu do obrotu zgodnie z zaleceniami producenta od dalszych badań klinicznych. To dwie różne sprawy. Jeśli lekarz kwalifikuje pacjenta do szczepienia w ramach ChPL, to wszystko jest w porządku. Nie jest to żaden eksperyment, lecz normalna procedura z użyciem zarejestrowanego wyrobu medycznego. Nie jest potrzebna na to ani szczególna zgoda pacjenta na eksperyment wymagana przepisami u.z.l., ani zgoda Komisji Bioetycznej.
Jednoczesne prowadzenie badań klinicznych różnych faz
W listopadzie 2020 Pfizer ogłosił zakończenie III fazy badania klinicznego szczepionki przeciw COVID-19, osiągając wszystkie pierwszorzędowe punkty końcowe w zakresie skuteczności. W grudniu 2020 szczepionka Comirnaty uzyskała warunkowe dopuszczenie do obrotu w UE. W ten sposób przeszła automatycznie do IV fazy badań klinicznych. Środowiska antyszczepionkowe postulują, że dopuszczone na rynek szczepionki są nieprzebadane i nie przeszły wszystkich wymaganych badań klinicznych. Na stronie szczepienia.pzh.gov.pl możemy przeczytać, że dopuszczenie do obrotu nastąpiło po ocenie wyników badania klinicznego III fazy potwierdzającego skuteczność i bezpieczeństwo szczepionki. Natomiast badania III fazy, które są kontynuowane, dotyczą m.in. oceny czasu utrzymywania się ochrony poszczepiennej.
Szczepionki przeciw COVID-19 są warunkowo dopuszczone do obrotu na podstawie wszystkich wymaganych badań laboratoryjnych, nieklinicznych na modelu zwierzęcym, badań klinicznych I i II fazy oraz okresowej oceny prowadzonych badań klinicznych najbardziej zaawansowanej trzeciej fazy badań. Pozwolenie jest wydawane gdy korzyści wynikające z natychmiastowej dostępności szczepionki dla pacjentów przewyższają ryzyko związane z faktem, że nie wszystkie dane z trwających badań 3 fazy są jeszcze dostępne. Szczepionki zostały dopuszczone po ocenie wyników badania klinicznego 3 fazy, gdzie potwierdzono skuteczność kliniczną i bezpieczeństwo szczepionki w ochronie przed objawami COVID-19. Badania kliniczne 3 fazy są jednak cały czas kontynuowane, aby ocenić takie parametry jak np. czas utrzymywania się ochrony poszczepiennej.
Zgodnie z zaleceniami ICH każde nowe zalecenie, nieznajdujące się w ChPL podlega oddzielnym badaniom i rejestracji. Dlatego równolegle do badań IV fazy prowadzi się również badania dla kolejnych wskazań jak np. stosowanie szczepionek przeciw COVID-19 u dzieci i kobiet ciężarnych. Prowadzone jest również badanie I fazy oceniające odpowiedź immunologiczną u pacjentów z HIV, oraz inne badanie I fazy dotyczące dzieci z ostrą białaczką szpikową i limfoblastyczną. W momencie zakończenia badań klinicznych dla nowego wskazania wydawane jest nowe dopuszczenie do obrotu i aktualizuje się ChPL i ulotkę.
Wykaz prowadzonych badań możemy znaleźć m.in. w europejskim rejestrze badań klinicznych prowadzonym przez EMA, oraz amerykańskim rejestrze prowadzonym przez NIH. Według nich szczepionka Comirnaty znajduje się obecnie jednocześnie w I, II, III i IV fazie badań klinicznych. Podobnie jak paracetamol, ibuprofen, aspiryna, witamina C i D oraz loperamid będący substancją aktywną popularnego leku na biegunkę.
Eksperymenty medyczne po myśli antyszczepionkowców
Dla porównania, amantadyna ogłoszona przez środowiska przeciwne szczepieniom jako przebadany i bezpieczny lek na COVID-19 znajduje się w tym momencie zarazem w II, III, jak i IV fazie badań klinicznych. Jednak zdecydowana większość z nich dotyczy jej zastosowania w chorobie Parkinsona. Dodatkowo nie została ona dopuszczona do stosowania w przypadku COVID-19 ani w trybie warunkowym, ani nawet w trybie awaryjnym. Jej wykorzystanie w tym zaleceniu jest ograniczone wyłącznie do badań klinicznych. Stąd można by pokusić się o stwierdzenie, że antyszczepionkowcy straszący „nieprzebadanymi i niebezpiecznymi szczepionkami” namawiają jednocześnie do stosowania nielegalnego eksperymentu medycznego, nieobjętego ubezpieczeniem, z użyciem nieprzebadanego produktu leczniczego, wbrew zasadom GCP.

Amantadyna – Święty Graal antyszczepionkowców
Jedna z czołowych ekspertek tego ruchu, psychiatra Katarzyna Ratkowska, powtarza niestrudzenie przy każdym wystąpieniu publicznym, że amantadyna jest stosowana od wielu lat i jej bezpieczeństwo jest bardzo dobrze znane. Nie można jej odmówić racji, bezpieczeństwo jej stosowania jest faktycznie całkiem dobrze znane i można je sprawdzić chociażby czytając ulotkę produktu. Poza dość długą listą interakcji, przeciwskazań i działań niepożądanych, możemy przeczytać poniższy fragment:
W badaniach na zwierzętach wykazano, że amantadyna podawana w dużych dawkach ma działanie embriotoksyczne i teratogenne. W przypadku stosowania amantadyny u kobiet w ciąży, donoszono zarówno o urodzeniu zdrowych noworodków jak i o powikłaniach ciąży oraz wadach wrodzonych u dzieci (wady układu sercowo-naczyniowego, krótsze kończyny). Ponieważ amantadyna stosowana w czasie ciąży może powodować ciężkie wady wrodzone nie należy jej stosować u kobiet ciężarnych oraz planujących ciążę. Amantadyna jest wydzielana do pokarmu kobiecego; nie stosować w okresie karmienia piersią.
Tutaj warto też dodać, że według Ratkowskiej, jednym z druzgocących argumentów na to, że szczepionki są niebezpieczne i nieprzebadane, jest to, że w dalszym ciągu są one w trakcie badań klinicznych. Ratkowska nie ukrywa jednocześnie, że stosuje amantadynę u swoich pacjentów z COVID-19, mimo iż nie jest ona dopuszczona do stosowania w tym wskazaniu i również jest w trakcje badań klinicznych.
Bardziej szczegółowo o amantadynie pisaliśmy w artykule pt. Nie ma dowodów, że amantadyna jest cudownym lekiem na COVID-19
Czynniki, które wpłynęły na przyspieszenie terminu dopuszczenia do obrotu
Podnoszonym często argumentem na brak zaufania do szczepionek przeciwko COVID-19 jest ekspresowe tempo, w jakim one powstały. Szczególnie w przypadku szczepionek mRNA, które w masowym użyciu pojawiły się po raz pierwszy w wyniku obecnej pandemii. Należy jednak mieć na uwadze, że technologia wykorzystana w ich produkcji nie jest całkowicie nowa. Podwaliny pod jej rozwój powstały już we wczesnych latach 90′, jednak dopiero osiągnięcia biotechnologii ostatnich lat pozwoliły na ostateczne opracowanie platformy mRNA.
Dotychczasowe prace nad szczepionkami genetycznymi
Wcześniejsze prace obejmowały badania nad szczepionkami przeciwko wściekliźnie, wirusowi HIV, Zika i grypy. W trakcie opracowania były również szczepionki przeciwko SARS i MERS, jednak te były szczepionkami DNA i nie weszły ostatecznie do użytku. Największe jednak nadzieje pokłada się w szeroko zakrojonych badaniach nad wykorzystaniem szczepionek mRNA w onkologii. Istnieje bardzo realna perspektywa produkcji szczepionek spersonalizowanych. Jest to osiągalne dzięki analizie sekwencji genetycznej nowotworu oraz możliwości szybkiego wytworzenia szczepionki mRNA ukierunkowanej na konkretny antygen nowotworowy specyficzny dla danego pacjenta.

Moce przerobowe, finansowanie i przepływ informacji
Kolejnym czynnikiem wpływającym na tempo opracowania szczepionek było zaangażowanie licznych ośrodków z całego świata, których prace skupiły się na palącym problemie pandemii. Badania nad SARS-CoV-2 były priorytetem, więc nie szczędzono na finansowaniu, stąd naukowcy mieli niemal nieograniczone możliwości i mogli badać wiele hipotez jednocześnie. Został również upłynniony przepływ informacji, dzięki temu, że czasopisma naukowe zdecydowały się na udostępnianie artykułów dotyczących SARS-CoV-2 poza pay-wall’em i często jeszcze przed publikacją, a nawet recenzją, w formie preprintów.
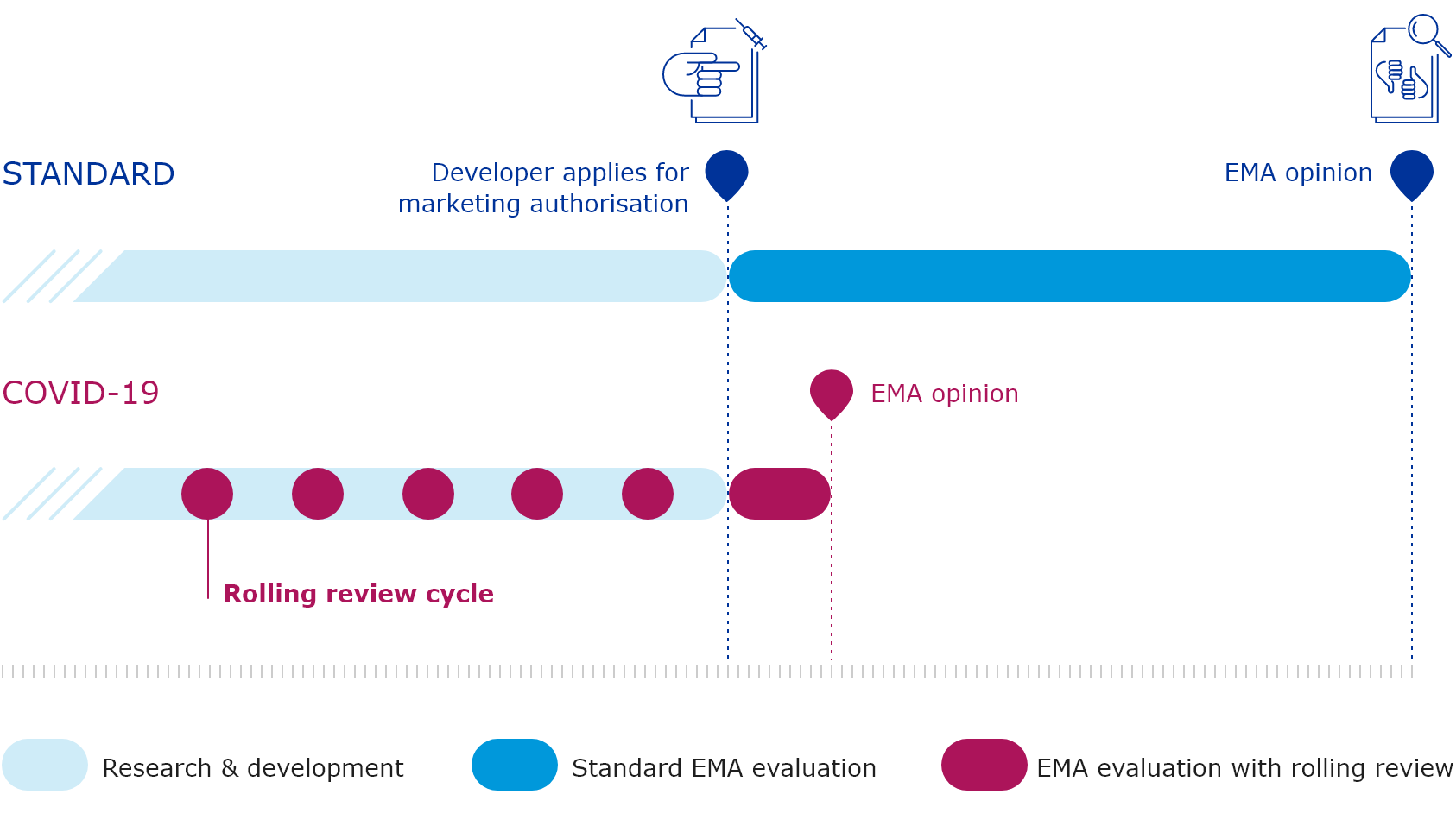
Przegląd kroczący
Poczyniono również kroki administracyjne by maksymalnie skrócić czas potrzebny na dopięcie formalności niezbędnych do dopuszczenia do obrotu. W ocenie badań klinicznych EMA zastosowała procedurę przeglądu kroczącego (przegląd etapowy, ang. rolling review). Jest on stosowany w celu przyspieszenia oceny obiecującego leku w sytuacji zagrożenia zdrowia publicznego, takiego jak trwająca pandemia. W standardowych warunkach, wszystkie dane popierające wniosek o dopuszczenie do obrotu produktu leczniczego muszą być złożone na początku procedury. W przypadku przeglądu kroczącego, dane są oceniane w miarę ich dostępności.
Warunkowe dopuszczenie do obrotu
Katarzyna Ratkowska zarzuca też często, że szczepionki zostały dopuszczone do obrotu w trybie awaryjnym. Nie do końca wiadomo, czy stosuje ten zabieg celowo w celu eskalacji strachu, czy ze względu na niezrozumienie działań administracyjnych związanych z wydawaniem takich zezwoleń. Jak wspomniano wcześniej w krajach europejskich szczepienia przeciw COVID-19 funkcjonują na zasadzie warunkowego dopuszczenia do obrotu. W Stanach Zjednoczonych wprowadzone one zostały natomiast w procedurze nadzwyczajnego pozwolenia na stosowanie (Emergency Use Authorization).
Warunkowe pozwolenie na dopuszczenie do obrotu (CMA, Conditional Marketing Authorization) – gwarantuje, że nadzór nad bezpieczeństwem farmakoterapii, wszelkie kontrole wytwarzania, w tym kontrole serii szczepionek, i inne obowiązki po wydaniu pozwolenia mają zastosowanie w prawnie wiążący sposób i są poddawane ciągłej ocenie przez komitety naukowe EMA. Tryb ten oznacza bardziej szczegółowy nadzór nad bezpieczeństwem i skutecznością szczepionek.
Nadzwyczajne pozwolenie na stosowanie (EUA, Emergency Use Authorization) – nie jest pozwoleniem na dopuszczenie do obrotu szczepionki, lecz pozwoleniem na tymczasowe stosowanie niedopuszczonej szczepionki. Umożliwia ono tymczasowe jej stosowanie w określonych warunkach, o ile zachodzą okoliczności nadzwyczajne.
Adwokat dr Monika Strus-Wołos na łamach pisma Palestra tłumaczy, że:
Warunkowość dotyczy jednak tylko dodatkowych obowiązków raportowania. Natomiast przed wydaniem pozwolenia przez Komisję Europejską odbyły się wszystkie trzy fazy badań klinicznych, po czym nastąpiła ocena przez EMA, dokładnie tak samo, jak dzieje się to w wypadku nieprzyspieszonego zatwierdzania szczepionek czy leków.
23 sierpnia 2021 r. amerykańska Agencja Żywności i Leków (FDA) przyznała pełną autoryzację dla szczepionki Pfizer-BioNTech przeciwko COVID-19 dla osób powyżej 16 roku życia. W USA szczepionka ta pozostaje również nadal dostępna na podstawie nadzwyczajnego pozwolenia na stosowanie (EUA) dla osób w wieku 12-15 lat oraz w celu trzeciej dawki u osób z obniżoną odpornością.
Odpowiedzialność wobec poszkodowanych
Jak możemy przeczytać na stronie Komisji Europejskiej:
Zgodnie z unijnym warunkowym pozwoleniem na dopuszczenie do obrotu odpowiedzialność spoczywa na posiadaczu pozwolenia na dopuszczenie do obrotu. Posiadacz pozwolenia na dopuszczenie do obrotu będzie odpowiedzialny za produkt i jego bezpieczne stosowanie. Warunkowe pozwolenie na dopuszczenie do obrotu jest ważne przez rok, odnawialne i przewiduje takie same prawa i taką samą odpowiedzialność posiadacza jak w przypadku standardowego pozwolenia na dopuszczenie do obrotu. Ponadto posiadacz warunkowego pozwolenia na dopuszczenie do obrotu ma szczególne obowiązki, takie jak ukończenie lub przeprowadzenie nowych badań w określonym terminie w celu potwierdzenia, że stosunek korzyści do ryzyka pozostaje dodatni.
Natomiast w przypadku nadzwyczajnego pozwolenia na stosowanie UE nakłada na państwa członkowskie obowiązek usunięcia odpowiedzialności administracyjnej i cywilnej producenta i posiadacza pozwolenia na dopuszczenie do obrotu.
Umowa o przejęciu odpowiedzialności producenta
W lutym 2021 Strus-Wołos i Wołos Kancelaria Adwokacka s.c. poinformowała o istnieniu umów z producentami szczepionek przeciwko COVID-19, w których odpowiedzialność za szkody związane z używaniem lub rozlokowaniem szczepionki, w tym odszkodowania, które normalnie poniósłby producent zostały przeniesione na kraje członkowskie UE.
Wspomniana kancelaria tłumaczy w innym wpisie, że nie oznacza to, że producent jest zwolniony z odpowiedzialności wobec poszkodowanego:
Taka umowa nic nie zmienia dla pacjenta. Pacjent nadal może pozwać wprost producenta (lub producenta oraz jednocześnie inne podmioty, w tym Skarb Państwa). Natomiast umowa przewiduje, że w razie wystąpienia przez poszkodowanego przeciwko producentowi z pozwem o odszkodowanie za szkodę na osobie lub szkodę na mieniu (jak np. koszty leczenia, utrata zarobków), producent w określonym terminie zawiadamia państwo, które na piśmie potwierdza, że w razie przegranej producenta pokryje te szkody. Umowa przewiduje alternatywnie, że jeżeli prawo danego państwa członkowskiego przewiduje taką możliwość, państwo może wstąpić do procesu w miejsce pozwanego producenta, za zgodą strony powodowej, tj. poszkodowanego pacjenta. Czyli pozywamy producenta, a jeżeli sprawa jest wygrana, pieniądze otrzymamy od Skarbu Państwa. Układ idealny, bo państwo nigdy nie może zbankrutować.
Podsumowanie
Teza jakoby narodowy program szczepień przeciwko COVID-19 był eksperymentem medycznym nie ma potwierdzenia w faktach. Rozpowszechnianie takich twierdzeń świadczy co najwyżej o braku elementarnej wiedzy na temat sposobu prowadzenia badań klinicznych i dopuszczania nowych produktów leczniczych na rynek. Świadome używanie takiego fałszywego argumentu przez osoby z wykształceniem medycznym świadczyć by mogło o celowej eskalacji poczucia strachu i negatywnych emocji wśród osób nieprzekonanych i przeciwnych szczepieniom lub o zupełnej niekompetencji do pełnienia zawodu lekarza. Jednym z zobowiązań Przyrzeczenia Lekarskiego, będącego preambułą do Kodeksu Etyki Lekarskiej, jest nie tylko służenie zdrowiu ludzkiemu i zapobieganie chorobom, ale również stałe poszerzanie swojej wiedzy. Rozpowszechnianie przez lekarzy fałszywych treści na temat szczepień jest złamaniem tego przyrzeczenia i naruszeniem zasad etyki lekarskiej.
Kodeks Etyki Lekarskiej:
Art. 56. 1. Powinnością każdego lekarza jest stałe uzupełnianie i doskonalenie swej wiedzy i umiejętności zawodowych, a także przekazywanie ich swoim współpracownikom.
Art.57.1. Lekarzowi nie wolno posługiwać się metodami uznanymi przez naukę za szkodliwe, bezwartościowe lub nie zweryfikowanymi naukowo. Nie wolno mu także współdziałać z osobami zajmującymi się leczeniem, a nie posiadającymi do tego uprawnień.
Art. 71. Lekarz ma obowiązek zwracania uwagi społeczeństwa, władz i każdego pacjenta na znaczenie ochrony zdrowia, a także na zagrożenie ekologiczne. Swoim postępowaniem, również poza pracą zawodową, lekarz nie może propagować postaw antyzdrowotnych.

Źródła
Ustawa o zawodach lekarza i dentysty: https://isap.sejm.gov.pl/isap.nsf/download.xsp/WDU19970280152/U/D19970152Lj.pdf
Prawo farmaceutyczne: https://isap.sejm.gov.pl/isap.nsf/download.xsp/WDU20011261381/U/D20011381Lj.pdf
Grzegorz Cessak: http://www.urpl.gov.pl/pl/informacja-prezesa-urz%C4%99du-z-dnia-6-sierpnia-2021-roku-w-sprawie-zasad-prowadzenia-bada%C5%84-klinicznych
Walter M., Badania kliniczne ‐ organizacja, nadzór, monitorowanie, Oinpharma, Warszawa 2004
Badania kliniczne szczepionek: https://www.badaniaklinicznewpolsce.pl/o-badaniach-klinicznych/nowe-horyzonty-farmakoterapii/podstawy-badan-klinicznych-szczepionek/
GCP: https://www.gcppl.org.pl/Portals/2/advertisings/ICH_GCP_E6_R2_wersja_polska_FINAL.pdf
Strus-Wołos i Wołos Kancelaria Adwokacka s.c. (z 10.01.2021) : https://www.facebook.com/permalink.php?story_fbid=3256344877804958&id=352011064905035
Strus-Wołos i Wołos Kancelaria Adwokacka s.c. (z 16.01.2021): https://www.facebook.com/permalink.php?story_fbid=3271382686301177&id=352011064905035
Pfizer zakończenie III fazy: https://www.pfizer.com.pl/o-firmie/press-room/pfizer-i-biontech-zakonczyly-faze-3-badania-klinicznego-potencjalnej-szczepionki
Pfizer dopuszeczenie do obrotu: https://www.pfizer.com.pl/o-firmie/press-room/pfizer-i-biontech-uzyskuja-dopuszczenie-do-obrotu-w-Unii-Europejskiej-szczepionki
szczepienia.pzh.gov.pl: https://szczepienia.pzh.gov.pl/faq/na-czym-polega-warunkowe-dopuszczenie-do-obrotu-szczepionek-przeciw-covid-19-w-europejskiej-agencji-lekow/
rejestr badań klinicznych (EU): https://www.clinicaltrialsregister.eu/ctr-search/search
rejestr badań klinicznych (USA): https://clinicaltrials.gov
bezpieczeństwo stosowania amantadyny: https://www.mp.pl/pacjent/leki/subst.html?id=4509
historia rozwoju szczepionek mRNA: https://www.sciencedirect.com/science/article/abs/pii/S1748013219301483
wykorzystanie szczepionek mRNA: https://www.mdpi.com/2076-393X/9/5/486/htm
wykorzystanie szczepionek mRNA: https://www.nature.com/articles/nrd.2017.243
SARS i MERS: https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-020-00695-2
Szczepionki MRNA w onkologii: https://link.springer.com/chapter/10.1007/978-3-319-42934-2_5
Komisja Europejska: https://ec.europa.eu/commission/presscorner/detail/pl/QANDA_20_2390
Strus-Wołos i Wołos Kancelaria Adwokacka s.c. (z 11.01.2021): https://www.facebook.com/permalink.php?story_fbid=3259875727451873&id=352011064905035
Strus-Wołos i Wołos Kancelaria Adwokacka s.c. (z 19.01.2021): https://www.facebook.com/permalink.php?story_fbid=3280965865342859&id=352011064905035
Strus-Wołos i Wołos Kancelaria Adwokacka s.c. (z 01.02.2021): https://www.facebook.com/permalink.php?story_fbid=3313065888799523&id=352011064905035
Kodeks Etyki Lekarskiej: https://nil.org.pl/uploaded_images/1574857770_kodeks-etyki-lekarskiej.pdf
FDA (23.08.2021): https://www.fda.gov/news-events/press-announcements/fda-approves-first-covid-19-vaccine